Condições
Quando se precisa de acesso direto ao essencial
Qt longo congênito
Descrição
Doença hereditária que compreende várias mutações genéticas distintas. Clinicamente se manifesta sob duas formas: Síndrome de Jervell e Lange-Nielsen (Qt longo associado à surdez congênita) e Síndrome de Romano-Ward (audição preservada). Foram identificadas diversas mutações que configuram os tipos de QT longo (LQT1, LQT2, LQT3 e etc). Atualmente, são registrados ao menos 13 tipos de QT longo (Tabela 1), mas se sabe que a maioria absoluta dos casos com identificação genética positiva se encontra dos tipos LQT1 (40-55%), LQT2(30-45%) e LQT3(5-10%).
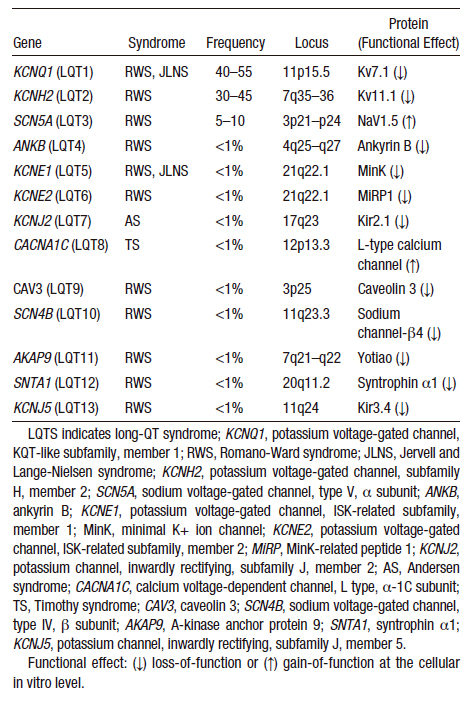
Schwartz et al / Circ Arrhythm Electrophysiol. 2012;5:868-877
O mecanismo comum envolvido com estas alterações são modificações do funcionamento dos canais iônicos da membrana celular e, consequentemente, mudanças heterogêneas do potencial de ação de diferentes regiões do miocárdio. Isto resulta em dispersão da refratariedade ventricular com propensão para arritmias com reentrada de fase 2 (pano de fundo para taquicardias ventriculares polimórficas).
Exemplos:
LQT1: diminuição da função da corrente Iks
LQT2: diminuição da função da corrente Ikr
LQT3: ganho de função da corrente de Na+
A incidência descrita na população geral é de
1 caso em 2000 a 2500 indivíduos.
Manifestações
A principal manifestação é a síncope, geralmente precedida por estresse físico ou emocional. A manifestação mais temida é a morte súbita cardíaca (habitualmente causada por degeneração de taquicardia polimórfica em fibrilação ventricular). Raramente a síndrome se manifesta com desenvolvimento de bloqueio atrioventricular. Dependendo do tipo genético envolvido, há uma predileção para certos gatilhos dos eventos.
LQT1: relação com atividade física ou estresse psicológico
LQT2: relação com estresse emocional (muitas vezes causado por estímulo auditivo intenso)
LQT3: relação com repouso e com sono
Diagnóstico
O diagnóstico clinico envolve a utlização do score de Schwartz (Tabela 2) publicado inicialmente em 1985 e submetido a diversas atualizações desde então.
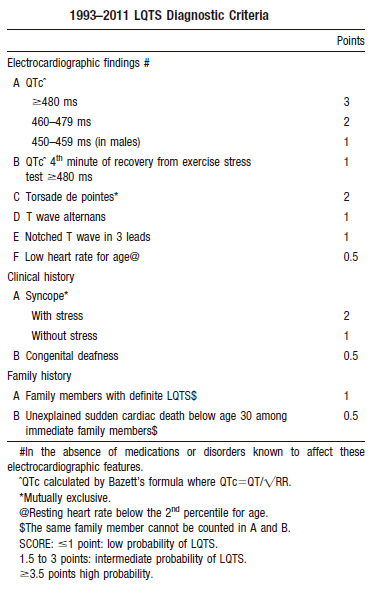
Schwartz and Crotti / Circulation. 2011;124:2181-2184
Métodos complementares envolvidos:
ECG
Fundamental para a mensuração do intervalo QTc (Figura demonstrando intervalo QT e fórmula para cálculo)
Apesar de não ser ter uma correção uniforme para todas as frequências cardíacas, o a fórmula para cálculo do QTc mais utilizado é a de Bazett. Além do prolongamento do intervalo QT, alguns pacientes apresentam outras aberrações como alternância de T, onda T bífida, onda T estreita e tardia.
QTc correção do intervalo QT de um dado paciente ajustando seu QT absoluto para uma frequência cardíaca de 60bpm.
HOLTER 24h
Em muitos casos o ECG de repouso pode não demonstrar prolongamento patológico do intervalo QT. No entanto, vários pacientes demonstram um comportamento dinâmico da repolarização ventricular.
Deste modo podemos flagrar variações importantes da duração e da forma do intervalo QT em alguns pacientes através de monitoração de 24h.
Teste ergométrico
Além da mensuração do intervalo QT na fase de recuperação, algumas vezes, há evidente falha no encurtamento do QTc durante a estimulação adrenérgica do esforço (em especial nos pacientes portadores de LQT1).
Ecocardiograma
Apesar de serem descritas discretas alterações no padrão de contratilidade dos portadores de QT longo congênito. O ecocardiograma é essencial para avaliação da função sistólica e para o diagnóstico diferencial de outras condições passíveis de aumento do intervalo QT como miocardite e isquemia.
Bioquímica
São fundamentais as mensurações sérias de K, Ca e Mg (frequentes causadores de perturbações do intervalo QT em pacientes críticos) e da função tireoideana (hipotireoidismo como eventual fator envolvido)
Diagnóstico molecular
Método ainda pouco utilizado em nosso meio. Mesmo que o diagnóstico clínico seja feito de forma razoavelmente segura, o teste genético deveria ser sempre empregado. Uma identificaçao positiva (~75%) nos obriga a realizar o "screening" familiar mesmo que parentes sejam assintomáticos e com ECG NORMAL (busca de portador assintomático).
Tratamento
O tratamento da síndrome do QT longo congênito depende largamente da sua apresentação clínica e envolve medidas não farmacológicas, drogas e intervenções cirúrgicas.
1) Evitar drogas que possam causar prolongamento do intervalo QT em todos os pacientes (www.azcert.org)
2) Evitar estresse físico e emocional
Afastar de atividade física intensa em todos os pacientes portadores de LQT1 (especialmente natação)
Atividade física recreacional pode ser eventualmente liberada em portadores de LQT2 e LQT3
Retirar telefone, despertador e outros dispositivos que possam ser fonte de ruído do quarto do paciente (especialmente no LQT2)
3) Tratamento medicamentoso
O uso de betabloqueador deve ser instituído em todos os pacientes sintomáticos que o tolerem
Seu uso deve ser fortemente considerado nos pacientes assintomáticos
(13% de primeira manifestação da síndrome é a morte súbita)
Existe forte benefício para os portadores de LQT1
Há benefício comprovado, mesmo que em menor escala, nos portadores de LQTS 2 e 3
Podem ser motivos para suspensão: asma e bradicardia significativas
Drogas de escolha: propranolol (2-3mg/kg/dia) e nadolol(1.5mg/kg/dia)
Atenolol e metoprolol têm índice de falha muito superior aos do propranolol e do nadolol,
portanto não devem ser consideradas drogas de primeira escolha
Em pacientes com LQT3, o uso de mexiletine (após dose de teste) pode levar a encurtamento significativo do intervalo QT e, assim, pode se mostrar útil associado ao betabloqueador
Suplementação de potássio em casos de perdas agudas por diarreia e vômitos podem ser indicadas para manter nível sério acima de 4mEq/l.
5) Simpatectomia
A simpatectomia esquerda se mostra útil na diminuição de
terapias apropriadas em portadores da síndrome que possuem Cardiodesfibrilador Interno (CDI)
Deve ser cogitada em crianças sintomáticas apesar do uso de betabloqueadores
ou intolerantes aos mesmos e que sejam muito pequenas para receber um CDI.
6) Dispositivos elétricos
Marca-passo definitivo: raramente implantado.
Deve ser considerado naquele doente que possui arritmia desencadeada por pausa.
Muitos autores consideram que, quando há indicação de implante de sistema de estimulação, seja implantando CDI para utilização como suporte tanto para bradicardia e pausas, como para terapia automática
de arritmias ventriculares sustentadas.
Desfibriladores internos: têm indicação nos sobreviventes de morte súbita (mesmo que virgens de tratamento medicamentoso) e nos pacientes sintomáticos apesar de doses apropriadas de betabloqueadores.
Figuras
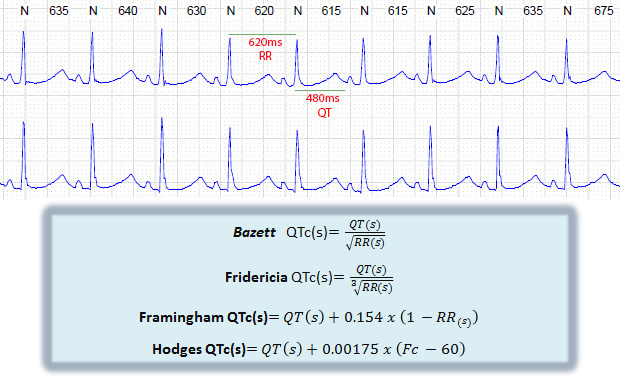
QT(ms): medida QT absoluto (medida do início do QRS ao fim da onda T)
RR(ms): medida do intervalo RR que antecede o batimento
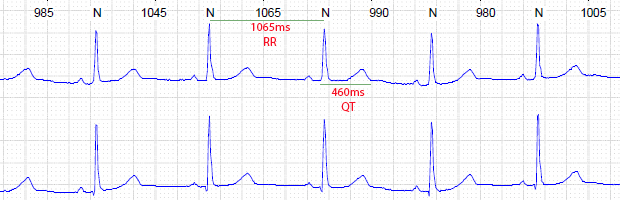
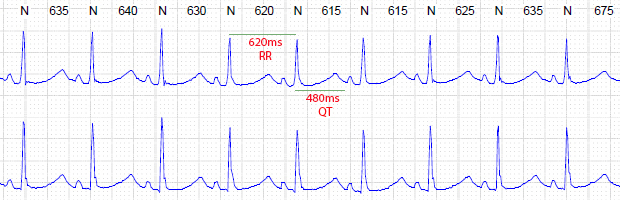
Figuras B e C. Exemplo variação de intervalo QT
em momentos distintos de holter de paciente com queixa de síncope
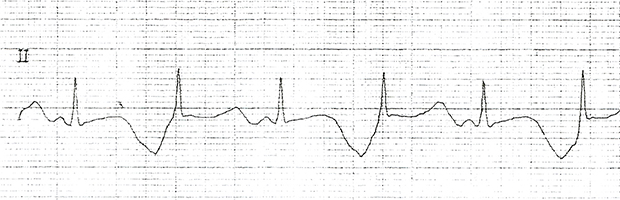
Paciente feminina, 16 anos. Portadora de surdez congênita e recuperada de fibrilação ventricular
Diagnóstico clínico de Síndrome de Jervell e Lange-Nielsen